 Welcome
Welcome
“May all be happy, may all be healed, may all be at peace and may no one ever suffer."
Congenital adrenal hyperplasia


Congenital adrenal hyperplasia (CAH) is a group of genetic disorders that affect the adrenal glands, which produce hormones that regulate various body functions. In CAH, the adrenal glands produce abnormal amounts of certain hormones, leading to a range of symptoms such as abnormal genital development, dehydration, electrolyte imbalances, and in severe cases, life-threatening adrenal crises. The most common type of CAH is caused by a deficiency of the enzyme 21-hydroxylase, which leads to a decrease in the production of cortisol and aldosterone and an increase in androgens. Treatment for CAH involves lifelong hormone replacement therapy to replace the missing hormones and to prevent the overproduction of androgens. In some cases, surgery may be needed to correct genital abnormalities. With proper treatment and management, most individuals with CAH can lead normal, healthy lives.
Research Papers
Disease Signs and Symptoms
Disease Causes
Congenital adrenal hyperplasia
The most common cause of CAH is the lack of the enzyme known as 21-hydroxylase. CAH may sometimes be called 21-hydroxylase deficiency. There are other much rarer enzyme deficiencies that also cause CAH.
Children who have the condition have two parents who either have CAH themselves or who are both carriers of the genetic mutation that causes the condition. This is known as the autosomal recessive inheritance pattern.
Disease Prevents
Congenital adrenal hyperplasia
There is no known way to prevent congenital adrenal hyperplasia. If you're thinking of starting a family and you're at risk of having a child with CAH, your doctor may recommend that you see a genetic counselor.
Disease Treatments
Your doctor will likely refer your child to a doctor who specializes in childhood hormonal issues (pediatric endocrinologist) for treatment of CAH. The health care team may also include other specialists, such as urologists, psychologists and geneticists.
Medications
The goal of treating CAH with medications is to reduce excess androgen production and replace deficient hormones. People who have the classic form of CAH can successfully manage the condition by taking hormone replacement medications throughout their lives. People who have nonclassic CAH may not require treatment or may need only small doses of corticosteroids.
Medications for CAH are taken on a daily basis. During periods of illness or significant stress, such as surgery, additional medications or higher doses may be needed.
Medications may include:
- Corticosteroids to replace cortisol
- Mineralocorticoids to replace aldosterone to help retain salt and get rid of excess potassium
- Salt supplements to help retain salt
Monitoring the effectiveness of medication includes regularly scheduled:
- Physical exams. The doctor will check your child's growth and development, including monitoring changes in height, weight, blood pressure and bone growth.
- Monitoring for side effects. The doctor will monitor your child for side effects, such as the loss of bone mass and impaired growth, particularly if steroid-type replacement medication doses are high and used long term.
- Blood tests to check hormone levels. It's critical to have regular blood tests to ensure hormone levels are balanced. A child who hasn't yet reached puberty needs enough cortisone to suppress androgens so that he or she can grow to a normal height. For females who have CAH, it's important to suppress androgens to minimize unwanted masculine characteristics. On the other hand, too much cortisone can cause Cushing syndrome.
Reconstructive surgery
In some female infants who have severe ambiguous genitalia as a result of classic CAH, doctors may recommend reconstructive surgery to improve genital function and make them look more feminine.
Surgery may involve reducing the size of the clitoris and reconstructing the vaginal opening. The surgery is typically performed between 2 and 6 months of age. Females who have reconstructive genital surgery may need more cosmetic surgery later in life.
Genital surgery is easier to perform when a child is very young. However, some parents choose to wait for surgery until their child is old enough to understand the risks and choose his or her own gender assignment.
Before making decisions about the best treatment approach for your child, talk with your doctor about these issues. Working together, you and your doctor can make informed choices that will help your child thrive.
Psychological support is important to the emotional health and social adjustment of female children who have genital abnormalities.
Prenatal treatment
Synthetic corticosteroids that cross the placenta to the fetus are controversial and considered experimental. More research is needed to determine the long-term safety and the effect of this treatment on fetal brain development.
Disease Diagnoses
Disease Allopathic Generics
Disease Ayurvedic Generics
Disease Homeopathic Generics
Disease yoga
Congenital adrenal hyperplasia and Learn More about Diseases
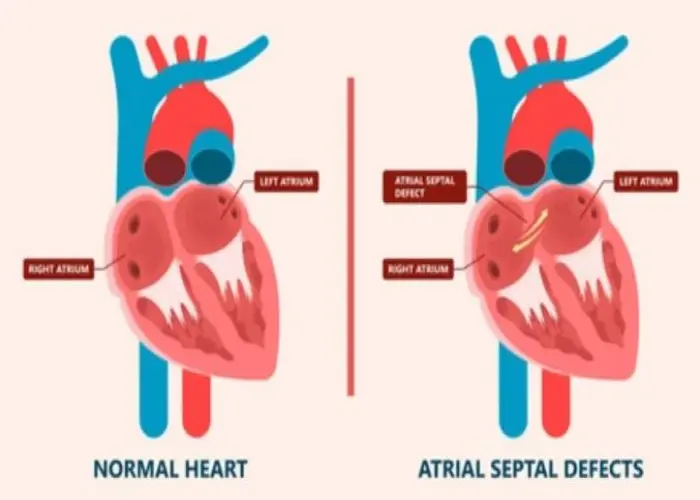
Atrial septal defect (ASD)
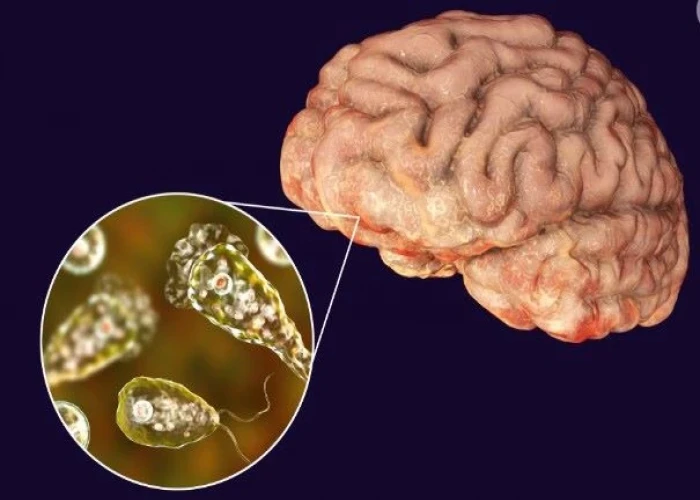
Naegleria infection

Selective IgA deficiency

Adjustment disorders
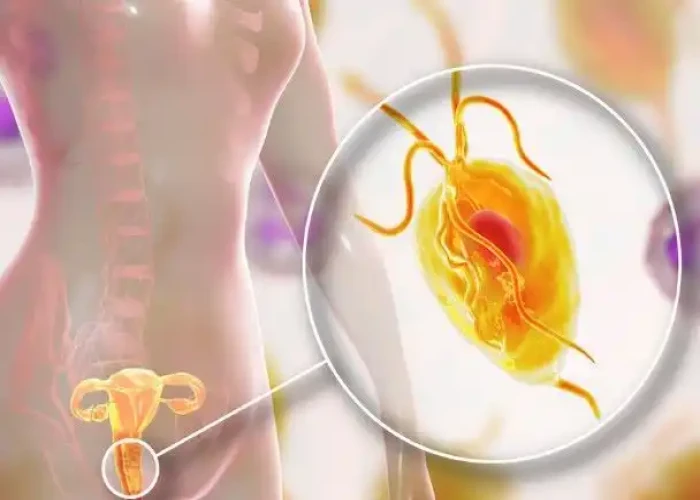
Trichomoniasis
Stroke

Keratitis

Syringomyelia
Congenital adrenal hyperplasia, CAH, Adrenal hyperplasia, জন্মগত অ্যাড্রিনাল হাইপারপ্লাজিয়া
To be happy, beautiful, healthy, wealthy, hale and long-lived stay with DM3S.