 Welcome
Welcome
“May all be happy, may all be healed, may all be at peace and may no one ever suffer."
DiGeorge syndrome (22q11.2 deletion syndrome)



DiGeorge syndrome, also known as 22q11.2 deletion syndrome, is a genetic disorder caused by a missing piece of chromosome 22. This condition affects the development of several body systems, including the heart, immune system, and facial features.
The symptoms of DiGeorge syndrome can vary widely, even among individuals with the same genetic mutation. Some common symptoms include congenital heart defects, cleft palate, learning disabilities, recurrent infections, and low levels of calcium in the blood. Individuals with DiGeorge syndrome may also have distinctive facial features, such as small ears, a small jaw, and widely spaced eyes.
Diagnosis of DiGeorge syndrome typically involves a physical exam, as well as genetic testing to confirm the deletion of the 22q11.2 region. In some cases, additional testing may be needed to assess the extent of organ involvement and to guide treatment.
Treatment of DiGeorge syndrome typically focuses on managing symptoms and complications associated with the condition. For example, congenital heart defects may require surgical repair, while infections may be treated with antibiotics. Individuals with low calcium levels may need calcium and vitamin D supplements.
In addition to medical treatment, individuals with DiGeorge syndrome may benefit from early intervention and educational services to help manage learning and behavioral challenges. Genetic counseling may also be recommended to help families understand the risks of passing the condition on to future generations.
In conclusion, DiGeorge syndrome is a genetic disorder caused by a missing piece of chromosome 22, and can affect multiple body systems. Symptoms can vary widely, but may include congenital heart defects, learning disabilities, recurrent infections, and low calcium levels. Treatment typically focuses on managing symptoms and complications, and may include surgical repair, medications, and early intervention and educational services. If you suspect that you or your child may have DiGeorge syndrome, it is important to seek medical attention to receive a proper diagnosis and develop an appropriate treatment plan.
Research Papers
Disease Signs and Symptoms
- Irregular heart sound (heart murmur)
- Blue skin (cyanosis)
- Frequent infections
- Delayed growth and development
- Difficulty breathing (dyspnea)
- Weak muscle tone (hypotonia)
- Delay in speaking
- Underdeveloped chin, low-set ears, wide-set eyes or a narrow groove in the upper lip
Disease Causes
DiGeorge syndrome (22q11.2 deletion syndrome)
Each person has two copies of chromosome 22, one inherited from each parent. If a person has DiGeorge syndrome (22q11.2 deletion syndrome), one copy of chromosome 22 is missing a segment that includes an estimated 30 to 40 genes. Many of these genes haven't been clearly identified and aren't well-understood. The region of chromosome 22 that's deleted is known as 22q11.2.
The deletion of genes from chromosome 22 usually occurs as a random event in the father's sperm or in the mother's egg, or it may occur early during fetal development. Rarely, the deletion is an inherited condition passed to a child from a parent who also has deletions in chromosome 22 but may or may not have symptoms.
Disease Prevents
DiGeorge syndrome (22q11.2 deletion syndrome)
In some cases, DiGeorge syndrome (22q11.2 deletion syndrome) may be passed from an affected parent to a child. If you're concerned about a family history of 22q11.2 deletion syndrome, or if you already have a child with the syndrome, you may want to consult a doctor who specializes in genetic disorders (geneticist) or a genetic counselor for help in planning future pregnancies.
Disease Treatments
Although there is no cure for DiGeorge syndrome (22q11.2 deletion syndrome), treatments can usually correct critical problems, such as a heart defect or cleft palate. Other health issues and developmental, mental health or behavioral problems can be addressed or monitored as needed.
Treatments and therapy for 22q11.2 deletion syndrome may include interventions for:
- Hypoparathyroidism. Hypoparathyroidism can usually be managed with calcium supplements and vitamin D supplements.
- Heart defects. Most heart defects associated with 22q11.2 deletion syndrome require surgery soon after birth to repair the heart and improve the supply of oxygen-rich blood.
- Limited thymus gland function. If your child has some thymic function, infections may be frequent, but not necessarily severe. These infections — usually colds and ear infections — are generally treated as they would be in any child. Most children with limited thymic function follow the normal schedule of vaccines. For most children with moderate thymus impairment, immune system function improves with age.
- Severe thymus dysfunction. If the impairment of the thymus is severe or there's no thymus, your child is at risk of a number of severe infections. Treatment requires a transplant of thymus tissue, specialized cells from bone marrow or specialized disease-fighting blood cells.
- Cleft palate. A cleft palate or other abnormalities of the palate and lip can usually be surgically repaired.
- Overall development. Your child will likely benefit from a range of therapies, including speech therapy, occupational therapy and developmental therapy. In the United States, early intervention programs providing these types of therapy are usually available through a state or county health department.
- Mental health care. Treatment may be recommended if your child is later diagnosed with attention-deficit/hyperactivity disorder (ADHD), autism spectrum disorder, depression, or other mental health or behavioral disorders.
- Management of other conditions. These may include addressing feeding and growth issues, hearing or vision problems, and other medical conditions.
Health care team
Because 22q11.2 deletion syndrome can result in so many problems, several specialists will likely be involved in diagnosing specific conditions, recommending treatments and providing care. This team will evolve as your child's needs change. Specialists on your care team may include these professionals and others, as needed:
- Children's health specialist (pediatrician)
- Expert in inherited disorders (geneticist)
- Heart specialist (cardiologist)
- Immune system specialist (immunologist)
- Ear, nose and throat (ENT) specialist
- Infectious disease specialist
- Hormone disorder specialist (endocrinologist)
- Surgeon who specializes in correcting such conditions as a cleft palate (oral and maxillofacial surgeon)
- Surgeon who specializes in correcting heart defects (cardiovascular surgeon)
- Occupational therapist to help develop practical, everyday skills
- Speech therapist to help improve verbal skills and articulation
- Developmental therapist to help develop age-appropriate behaviors, social skills and interpersonal skills
- Mental health professional, such as a pediatric psychiatrist or psychologist
Disease Diagnoses
Disease Allopathic Generics
Disease Ayurvedic Generics
Disease Homeopathic Generics
Disease yoga
DiGeorge syndrome (22q11.2 deletion syndrome) and Learn More about Diseases

Mammary duct ectasia
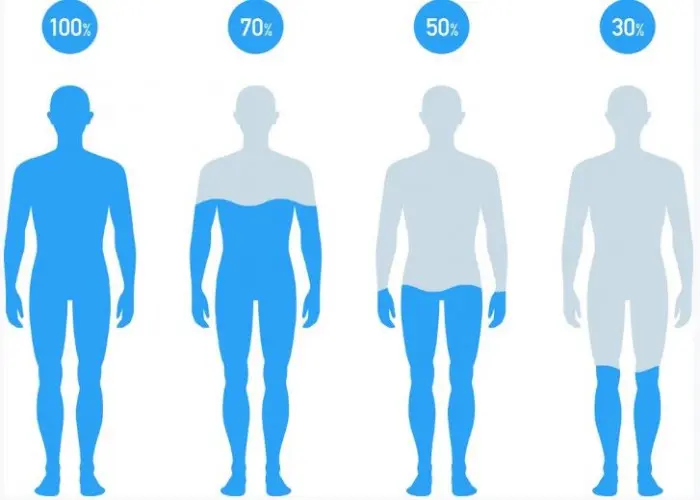
Dehydration
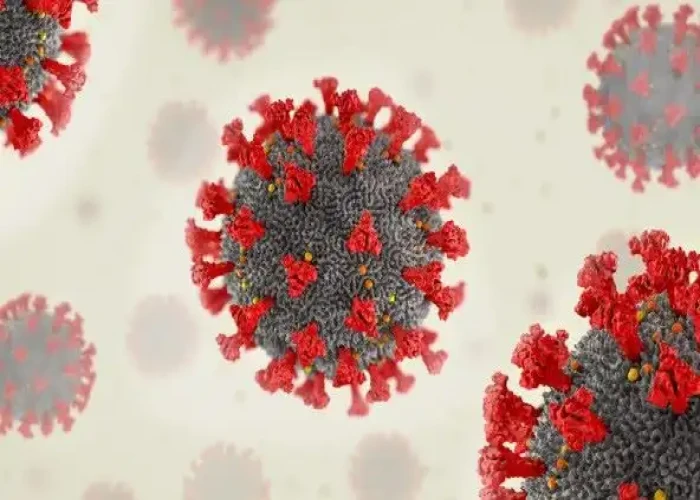
Severe acute respiratory syndrome (SARS)

Hoarding disorder
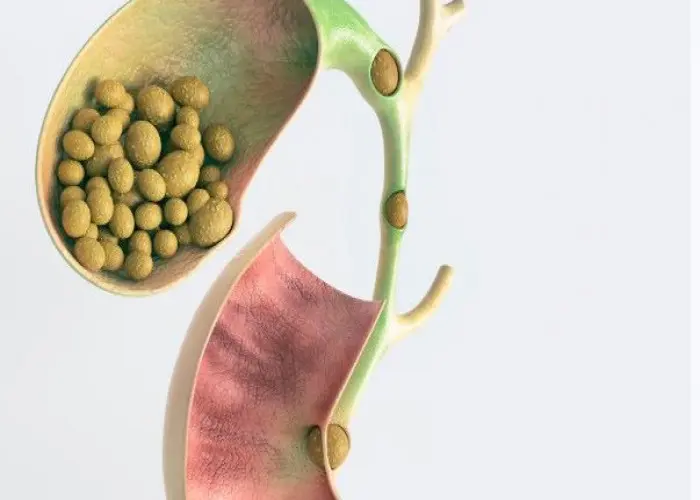
Gallstones

Peripheral nerve injuries

Zollinger-Ellison syndrome
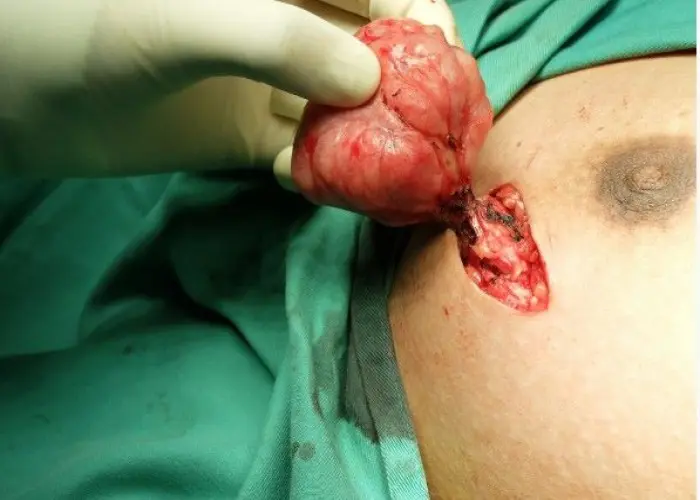
Fibroadenoma
digeorge syndrome, 22q11.2 deletion syndrome, ডিগার্জ সিন্ড্রোম, ২২কিউ১১.২ মুছে ফেলার সিন্ড্রোম
To be happy, beautiful, healthy, wealthy, hale and long-lived stay with DM3S.